
Introduction
Long QT syndrome (LQTS) is a disorder of ventricular myocardial repolarization characterized by a prolonged QT interval on ECG that can lead to symptomatic ventricular arrhythmias and an increased risk of sudden cardiac death (SCD).
The QT interval is the time from the start of the Q wave to the end of the T wave. QT interval represents the time taken for ventricular depolarisation and repolarisation.
The Long QT Syndrome (LQTS) is characterized on the ECG by prolongation of the heart rate corrected QT interval (QTc).
The corrected QT interval (QTc) estimates the QT interval at a standard heart rate of 60 bpm
- Bazett formula: QTc = QT / √ RR
- Fridericia formula: QTc = QT / RR 1/3
Overall, the average QTc in healthy persons during infancy is 400±20 milliseconds and increases slightly after puberty to 420±20 milliseconds.
Clinical diagnosis
- Corrected QT (QTc) ≥480 ms or a score >3
- In the presence of unexplained syncope, a QTc ≥460 ms is sufficient to make a diagnosis
- Irrespective of the QT duration if pathogenic LQTS station confirmed
Risc score calculator: https://www.mdcalc.com/tisdale-risk-score-qt-prolongation
Classification
LQTS may be either congenital or acquired.
- Congenital - mutations in at least 17 genes have been identified with congenital LQTS
- Mutations in KCNQ1 (LQT1 - swimming), KCNH2 (LQT2 – loud noise), and SCN5A (LQT3) account for approximately 80% of all congenital LQTS cases !!!
The subtypes of LQTS may be grouped into three categories:
- Autosomal dominant LQTS (Romano–Ward syndrome; prevalence 1 in 2500), which includes LQT1–6 and LQT9–13 and is characterized by an isolated prolongation of the QT interval
- Autosomal dominant LQTS with extracardiac manifestation, comprising:
- LQT7 (Andersen–Tawil syndrome), which shows a prolonged QT interval with prominent U wave, polymorphic or bidirectional VT facial dysmorphisms and hyper-/hypokalemic periodic paralysis
- LQT8 (Timothy syndrome), characterized by prolonged QT, syndactyly, cardiac malformations, autism spectrum disorder and dysmorphisms
- Autosomal recessive LQTS (Jervell and Lange–Nielsen syndrome), which combines an extremely prolonged QT interval with congenital deafness.
- Acquired
- Medication
- Antiarrhythmic drugs (sotalol, amiodarone, propafenone, flecainide, etc)
- Antibiotics (eg, macrolide and fluoroquinolone antibiotics, some antifungal and antiviral drugs, etc.)
- Psychotropic medications (chlorpromazine, haloperidol, mesoridazine, etc.)
- Antidepressants (SSRI)
- Gastric motility agents (eg, cisaprid)
More about drugs that are prolonging QT interval: https://www.crediblemeds.org/ - Female sex
- Hypokalemia, hypomagnesaemia, hypocalcaemia
- Bradycardia
- Hypothermia
- Myocardial ischemia
- Post-cardiac arrest
- Raised intracranial pressure
Treatment
- Lifestyle modification
- No competitive sports in all LQTS patients
- No swimming in LQT1 patients
- Avoid nightly noise in LQT2 patients (e.g. no alarm clock)
- Medication: beta-blockers can reduce the risk of sudden death in patients in whom a genetic defect has been found, but no QT prolongation is visible on the ECG.
- ICD implantation
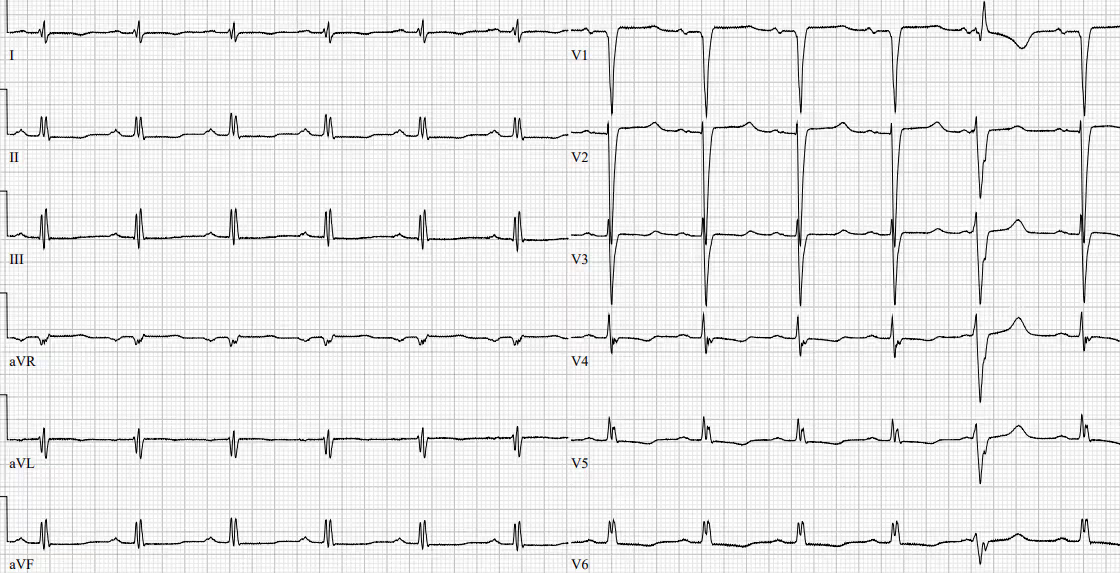
ECG 1 Patient with amiodarone associated LQT

ECG 2 Patient with amiodarone related LQT leading to ventricular ectopy

ECG 3 Patient with long QT after administration of amiodarone and tiaprid.
Torsade de Pointes (TdP)
Is a form of polymorphic ventricular tachycardia associated with a long QT interval on the resting ECG. TdP is characterized by morphology in which the QRS complexes “twist” around the isoelectric line (cycling of the QRS axis through 180 degrees every 5 to 20 beats).
It occurs in the setting of acquired or congenital QT interval prolongation and typically has a rate between 160 and 250 beats per minute.
Drug-related TdP ia caused by early afterdepolarizations and triggered activity resulting from prolonged repolarization.
Symptoms
TdP usually leads to hemodynamic instability and collapse. Moreover TdP can also degenerate into ventricular fibrillation.
Treatment
- Withdrawal of any QTc prolonging drugs
- Correction of electrolyte abnormalities (potassium repletion up to 4.5 to 5 mmol/liter).
- MgSO4 1-2 g i.v (decreases early afterdepolarizations)
- DC version
- Beta blockers as a prevention to patients with congenital LQT
- ICD implantation
ECG 4 Torsade de Pointes episode in a patient with drug induced LQT

ECG 5 R on T phenomenon leading to TdP in a patient with LQT

References
- Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez-Madrid A, Nikolaou N, Norekvål TM, Spaulding C, Van Veldhuisen DJ; Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Europace. 2015 Nov;17(11):1601-87. doi: 10.1093/europace/euv319. Epub 2015 Aug 29. PMID: 26318695.
- Long QT syndrome. UpToDate.
- https://en.ecgpedia.org/wiki/Long_QT_Syndrome
- https://litfl.com/qt-interval-ecg-library/
.jpg)
Introduction
Long QT syndrome (LQTS) is a disorder of ventricular myocardial repolarization characterized by a prolonged QT interval on ECG that can lead to symptomatic ventricular arrhythmias and an increased risk of sudden cardiac death (SCD).
The QT interval is the time from the start of the Q wave to the end of the T wave. QT interval represents the time taken for ventricular depolarisation and repolarisation.
The Long QT Syndrome (LQTS) is characterized on the ECG by prolongation of the heart rate corrected QT interval (QTc).
The corrected QT interval (QTc) estimates the QT interval at a standard heart rate of 60 bpm
- Bazett formula: QTc = QT / √ RR
- Fridericia formula: QTc = QT / RR 1/3
Overall, the average QTc in healthy persons during infancy is 400±20 milliseconds and increases slightly after puberty to 420±20 milliseconds.
Clinical diagnosis
- Corrected QT (QTc) ≥480 ms or a score >3
- In the presence of unexplained syncope, a QTc ≥460 ms is sufficient to make a diagnosis
- Irrespective of the QT duration if pathogenic LQTS station confirmed
Risc score calculator: https://www.mdcalc.com/tisdale-risk-score-qt-prolongation
Classification
LQTS may be either congenital or acquired.
- Congenital - mutations in at least 17 genes have been identified with congenital LQTS
- Mutations in KCNQ1 (LQT1 - swimming), KCNH2 (LQT2 – loud noise), and SCN5A (LQT3) account for approximately 80% of all congenital LQTS cases !!!
The subtypes of LQTS may be grouped into three categories:
- Autosomal dominant LQTS (Romano–Ward syndrome; prevalence 1 in 2500), which includes LQT1–6 and LQT9–13 and is characterized by an isolated prolongation of the QT interval
- Autosomal dominant LQTS with extracardiac manifestation, comprising:
- LQT7 (Andersen–Tawil syndrome), which shows a prolonged QT interval with prominent U wave, polymorphic or bidirectional VT facial dysmorphisms and hyper-/hypokalemic periodic paralysis
- LQT8 (Timothy syndrome), characterized by prolonged QT, syndactyly, cardiac malformations, autism spectrum disorder and dysmorphisms
- Autosomal recessive LQTS (Jervell and Lange–Nielsen syndrome), which combines an extremely prolonged QT interval with congenital deafness.
- Acquired
- Medication
- Antiarrhythmic drugs (sotalol, amiodarone, propafenone, flecainide, etc)
- Antibiotics (eg, macrolide and fluoroquinolone antibiotics, some antifungal and antiviral drugs, etc.)
- Psychotropic medications (chlorpromazine, haloperidol, mesoridazine, etc.)
- Antidepressants (SSRI)
- Gastric motility agents (eg, cisaprid)
More about drugs that are prolonging QT interval: https://www.crediblemeds.org/ - Female sex
- Hypokalemia, hypomagnesaemia, hypocalcaemia
- Bradycardia
- Hypothermia
- Myocardial ischemia
- Post-cardiac arrest
- Raised intracranial pressure
Treatment
- Lifestyle modification
- No competitive sports in all LQTS patients
- No swimming in LQT1 patients
- Avoid nightly noise in LQT2 patients (e.g. no alarm clock)
- Medication: beta-blockers can reduce the risk of sudden death in patients in whom a genetic defect has been found, but no QT prolongation is visible on the ECG.
- ICD implantation
ECG 1 Patient with amiodarone associated LQT
ECG 2 Patient with amiodarone related LQT leading to ventricular ectopy
ECG 3 Patient with long QT after administration of amiodarone and tiaprid.
Torsade de Pointes (TdP)
Is a form of polymorphic ventricular tachycardia associated with a long QT interval on the resting ECG. TdP is characterized by morphology in which the QRS complexes “twist” around the isoelectric line (cycling of the QRS axis through 180 degrees every 5 to 20 beats).
It occurs in the setting of acquired or congenital QT interval prolongation and typically has a rate between 160 and 250 beats per minute.
Drug-related TdP ia caused by early afterdepolarizations and triggered activity resulting from prolonged repolarization.
Symptoms
TdP usually leads to hemodynamic instability and collapse. Moreover TdP can also degenerate into ventricular fibrillation.
Treatment
- Withdrawal of any QTc prolonging drugs
- Correction of electrolyte abnormalities (potassium repletion up to 4.5 to 5 mmol/liter).
- MgSO4 1-2 g i.v (decreases early afterdepolarizations)
- DC version
- Beta blockers as a prevention to patients with congenital LQT
- ICD implantation
ECG 4 Torsade de Pointes episode in a patient with drug induced LQT
ECG 5 R on T phenomenon leading to TdP in a patient with LQT
References
- Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez-Madrid A, Nikolaou N, Norekvål TM, Spaulding C, Van Veldhuisen DJ; Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Europace. 2015 Nov;17(11):1601-87. doi: 10.1093/europace/euv319. Epub 2015 Aug 29. PMID: 26318695.
- Long QT syndrome. UpToDate.
- https://en.ecgpedia.org/wiki/Long_QT_Syndrome
- https://litfl.com/qt-interval-ecg-library/


