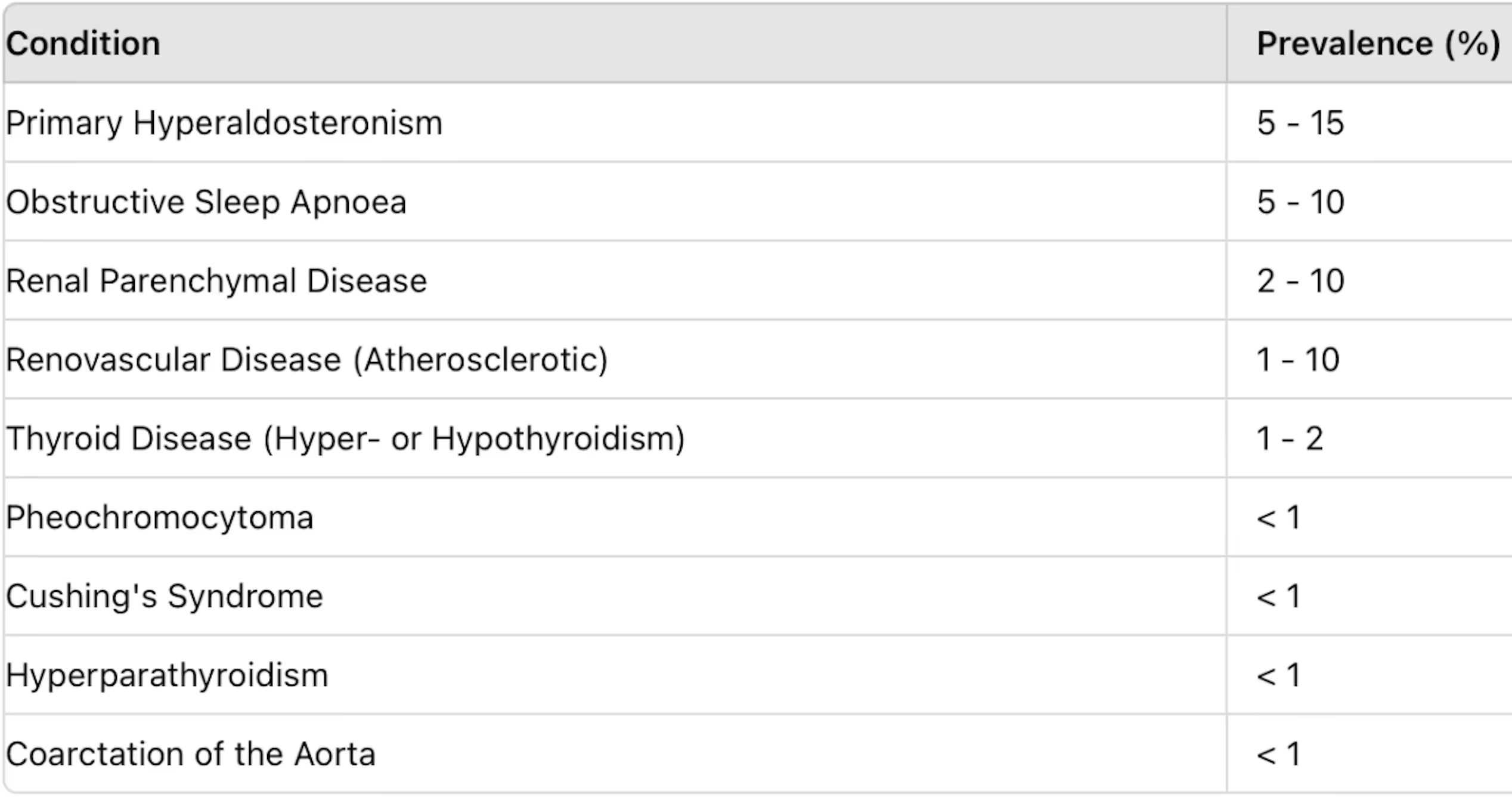
Junctional rhythm as a complication of Fabry Disease
A 54-year old patient with Fabry disease, mechanical valve prosthesis in mitral position, and history of kidney transplant was admitted for obstructive hydronephrosis and bradycardia with junctional rhythm (30-35/min).
Figure 1 ECG on admission with junctional rhythm

The mitral valve disease, newly diagnosed conduction abnormalities, as well as the chronic renal failure as the reason for kidney transplantation, have all been attributed to the patient’s primary inherited metabolic illness - Fabry disease.
Upon admission, TTE was performed and showed the following findings:
- Marked hypertrophy and echogenicity of the left ventricular myocardium (normal systolic function)
- Mitral valve mechanical prosthesis with stable gradients in comparison to previous examination, without significant regurgitation.
- Aortic valve thickened, with mild stenosis.
- Enormous left atrial dilatation (LAVi 194.1 cm3/m2).
- Severe tricuspid regurgitation, but hard to accurately quantify due to right atrial dilatation (63.00 cm2)
Video 1 TTE on admission

Video 2 Four chamber view on admission TTE

Figure 2 Severe biatrial dilatation on TTE

Despite isoprenaline therapy and discontinuation of bradycardia medication, junctional rhythm persisted. Given the good systolic function of both ventricles, the patient was indicated for single-chamber pacemaker implantation. With regards to the patient’s kidney drainage and thus heightened infection risk along with looming haemodialyzation therapy, MICRA (electrode-less) pacemaker was deemed the most suitable option.
Video 3 Skiascopy during MICRA insertion

The pacemaker was successfully implanted via right femoral vein to the apical portion of the right ventricular septum since the patient presented with midventricular obstruction of the left ventricular septum during the procedure.
The patient was then transferred back to the nephrology department for the follow-up treatment of hydronephrosis.
Clinical context
Fabry disease (FD) is an X-linked lysosomal storage disease (caused by a variety of mutations in the GLA gene on Xq22.1) with an incidence of about 1:120000, but mutation incidence is as high as 1:1250.
The cause of the classical variant stands the lack of alpha-galactosidase activity, which results in the accumulation of globotriaosylceramide (Gb3) in cells of various systems. This causes the disease’s wide-spread multi-systemic effects, namely paresthesias, skin and eye, cardiovascular, cerebrovascular and renal disorders.
The mean age of onset of the first symptom is before 10 years of age. Atypical variants of FD involve residual enzyme activity, resulting in milder, more often than not restricted to only one organ, symptoms with a later onset, usually between 40 and 60 years of age.
Undiagnosed patients with FD can be detected by screening in at-risk populations, such as patients with end-stage renal disease, unexplained myocardial hypertrophy or early stroke (18-55 years of age).
On the forefront of FD stands facial dysmorphism, also described as ‘Fabry facies’, characterized by periorbital fullness, bushy eyebrows, recessed forehead, pronounced nasal angle, bulbous nasal tip, full lips, broad alar base. It is possible that these features become more pronounced with age in regard to the accumulation of Gb3.
Figure 3 Fabry facies

Bica, Ríos & Elena, Blanca & Saldarriaga-Rivera, Lina & Maria, Lina & Breno, Valdetaro. (2014). Manifestaciones musculoesqueléticas de la Enfermedad de Fabry. 1. 360-364
The most common subjective symptom is neuropathic pain, with the average onset of about 15 years of age. It begins in the extremities and radiates proximally with the trigger being any physical or emotional stress.
Skin abnormalities, namely angiokeratomas, teleangiectasia and sweating abnormalities (an-/hypo-/hyper-hidrosis) are among the most commonly observed FD symptoms and hence lead to diagnoses being made in the dermatology departments. Another hallmark of FD are whorl-like opacities resulting from changes in the subepithelial layers of the cornea, also known as cornea verticillata.
Figure 5 Angiokeratomas

Moiseev S, Karovaikina E, Novikov PI, et al. (2020). What rheumatologist should know about Fabry disease: Annals of the Rheumatic Diseases 2020;79:e71.
Figure 5 Cornea verticillata

Lamont, Meon & Madge, Simon & Quinn, Anthony & Anderson, David & Habib, Nabil. (2010). Vortex keratopathy and fabry disease: a case series highlighting the role of the optometrist.
Cardiac involvement
Progressive cardio- and renovascular abnormalities are among the most common causes of death in all patients with FD. A so-called ‘Cardiac variant’ has also been recognized in a large number of patients, who present with a late-onset phenotype manifesting mostly as a left ventricular hypertrophy (LVH) or hypertrophic cardiomyopathy (HCM). The absolute majority of cardiovascular symptoms of FD begin manifesting between 30 and 40 years of age. The heart is the most susceptible tissue for Gb3 accumulation of the whole body and therefore presents with a broad spectrum of symptoms, among which angina pectoris, palpitations, syncope, dyspnoea and heart failure are the most notable.
Symmetrical progressive LVH is the predominant finding, diagnosed by imaging techniques (echocardiography, MRI) and ECG (hypertrophy voltage criteria), although a small portion (5%) of patients develop asymmetrical septal hypertrophy. While LV systolic function is usually within normal range, diastolic dysfunction is very typical. Increased oxygen demand from the hypertrophic muscle, along with endothelial infiltration and dysfunction causes significantly reduced coronary flow reserve. Although this leads to recurrent angina pectoris in a portion of patients, actual myocardial infarction is very rare in patients with FD and selective coronarography usually does not reveal coronary stenotic lesions.
Figure 6 ECG stripe showing significant left ventricular hypertrophy

Moiseev S, Karovaikina E, Novikov PI, et al. (2020). What rheumatologist should know about Fabry disease: Annals of the Rheumatic Diseases 2020;79:e71.
Figure 7 MRI showing diffuse left ventricular hypertrophy

Linhart A. The heart in Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Chapter 20. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11576/
Electrophysiological abnormalities are also very commonly involved in FD, on ECG typically with high voltage and short PR interval due to accelerated AV conduction or preexcitation (which occurs often among lysosomal storage diseases in general). Conduction system dysfunction; bundle branch and AV blocks of varying degrees, progressive sinus node dysfunctions occur, among others, with disease progression and very often require pacemaker implantation. Palpitation and arrhythmias, such as supraventricular tachycardia or atrial flutter and fibrillation accompany later stages of the disease.
Figure 8 Typical ECG of a patient with Fabry disease: short PR intervals, left ventricular hypertrophy and prominent ST depressions and T-wave inversions

Linhart A. The heart in Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Chapter 20. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11576/
Infiltrative changes within valvular fibroblasts cause also valvular involvement, most commonly resulting in mitral valve prolapse, usually with only mild to medium regurgitation, but occasionally requiring surgical valve replacement. Echocardiography usually reveals valvular thickening and deformation.
Figure 9 Echocardiogram showing mild thickening of the mitral valve leaflets (arrows), with also significant thickening of the septum

Linhart A. The heart in Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Chapter 20. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11576/
Figure 10 Colour doppler echocardiogram showing mild to moderate mitral regurgitation, with noticeable bilateral atrial dilatation

Linhart A. The heart in Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Chapter 20. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11576/
Among renovascular abnormalities, otherwise unexplained proteinuria/albuminuria are especially alarming. The disease manifests through isosthenuria with progressive deterioration of renal funcion to end-stage renal disease, which, unless counteracted by therapy, results in the death of the patient. Cerebrovascular involvement includes thrombosis, transient ischemic attacks, basilar artery ischemia and aneurysms, seizures, hemiplegia and other serious neurological symptoms. A neurologic phenotype including decreased overall motor performance has been also cited. Gastrointestinal and pulmonary abnormalities are also of common occurrence.
Due to the basic genetic principles, it was initially thought that Fabry disease is an illness mostly exclusive for the male population and females serve their role as carriers. Nevertheless, it was recently discovered that heterozygous females may (based on individual factors) experience milder forms of Fabry disease. They are usually subject to painful neuropathies and isolated skin lesions, although disturbances of corneal opacity have been observed in about 70-80% of otherwise asymptomatic heterozygous females.
Diagnosis
Figure 9 Fabry disease diagnosis algorithm

Linhart A, Germain DP, Olivotto I, Akhtar MM, Anastasakis A, Hughes D, Namdar M, Pieroni M, Hagège A, Cecchi F, Gimeno JR, Limongelli G, Elliott P. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur J Heart Fail. 2020 Jul;22(7):1076-1096.
The primary blood test for the diagnosis of Fabry disease assesses the activity of the alpha-galactosidase A enzyme. Another blood test, which detects the presence of a fatty substance called lyso-Gb3, also can be performed and may indicate the severity of the disease. Finally, the blood sample can be used for genetic testing that looks for mutations in the GLA gene and may help confirm Fabry disease.
Diagnosis of cardiovascular involvement
Electrocardiography usually shows:
- Shortened PR interval, repolarization abnormalities and signs of LVH are early ECG features which precede the development of structural abnormalities.
- LV mass at the upper limits of normal range.
- Voltage signs of LVH, strain pattern and T-wave inversion in precordial leads are virtually always present.
- Sinus bradycardia and progressive conduction disease
Echocardiography is the most useful method for diagnosing and monitoring FD-related cardiomyopathy.
- Typical finding is concentric LV remodelling or hypertrophy without resting LVOT obstruction. Asymmetrical hypertrophy of the septum is also possible, although rare.
- LVOT obstruction can be provoked by exercise or stress, putting classical HCM in the differential diagnosis.
- Progressive myocardial fibrosis may lead to hypokinetic or akinetic LV.
- Other typical features include papillary muscle hypertrophy and right ventricular thickening.
- LV EF is usually normal, but can be reduced in patients with extensive fibrosis.
- Diastolic function is normal in the early phases of cardiac involvement, but quickly deteriorates with transmitral flow and mitral annular tissue Doppler velocities becoming abnormal.
- Left atrial dilatation is very common.
- Mitral and aortic valves are often thickened, with mild-to-moderate regurgitation, or possibly mitral valve prolapse in some patients. Stenotic lesions are exceptional.
- Mild-to-moderate aortic dilatation of the bulbus/ascending aorta is frequently seen in advanced cases. The risk of aortic dissection is predicted to be very low.
Cardiac-MRI provides an accurate assessment of LV size, mass and geometry, gadolinium contrast enhancement can help visualize myocardial fibrosis.
Endymyocardial biopsy may be considered in some variants patients, high residual enzyme activity (>10%) or low lyso-Gb3 levels to confirm or exclude FD as the cause of LVH. It is however not a recommended method for treatment efficacy follow-up.
Electrophysiological study is recommended in patients with persistent or recurrent SVT to guide therapy.
- Should be also considered in patients with manifest pre-excitation or SA disease or AV block
- Presence of short PR intervals as an isolated finding is not an indication for an electrophysiological study
Laboratory tests aids detection of non-cardiac conditions that cause or exacerbate ventricular dysfunction.
- Also a critical part of monitoring renal function and progression of the renovascular involvement
- Elevated plasma inflammatory markers suggest disease progression
- Plasma lyso-Gb3 values have a significant role in monitoring therapy efficacy.
- Plasma NT-proBNP should be used for monitoring progression of cardiac involvement
Treatment
Standard, yet still fairly new treatment for FD is enzyme replacement therapy (ERT), which aims to supply a recombinant version of the genetically deficient enzyme. It has been shown to significantly alleviate the cardiac, renal, and neuropathic effects of FD. The enzyme is administered every 2 weeks. Novel therapy based on pharmacological chaperone is now available for FD patients of certain variants. Many studies have demonstrated a benefit in FD patients when ERT is initiated early.
Cardiologic treatment involves standard measures of reducing cardiovascular risk (e.g.: statins, antihypertensive therapy). Beta-blockers should be used only with great caution, as they have been proved to aggravate symptomatic bradycardia and AV conduction impairment in some patients - dihydropyridine Ca blockers are deemed the safer and more effective therapy. Antiaggregation in all patients with cardiac symptoms, anticoagulation in patients with supraventricular rhythm disturbances. Septal alcohol ablation can be also considered for patients with LVOT obstruction.8,9
Patients also profit from effective pain management. Prophylaxis of secondary complications is in the gestion of adequate dispensarisation. Other complications should be treated in accordance with protocols of the respective departments, with kidney transplant due to end-stage renal disease among the most significant.
References
- Chan B, Adam DN. A Review of Fabry Disease. Skin Therapy Lett. 2018 Mar;23(2):4-6. PMID: 29562089.
- Deegan PB, Bähner F, Barba M, et al. Fabry disease in females: clinical characteristics and effects of enzyme replacement therapy. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Chapter 30. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11591/
- Mehta A, Widmer U. Natural history of Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Chapter 19. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11572/
- Bica, Ríos & Elena, Blanca & Saldarriaga-Rivera, Lina & Maria, Lina & Breno, Valdetaro. (2014). Manifestaciones musculoesqueléticas de la Enfermedad de Fabry. 1. 360-364.
- Moiseev S, Karovaikina E, Novikov PI, et al. (2020). What rheumatologist should know about Fabry disease: Annals of the Rheumatic Diseases 2020;79:e71.
- Lamont, Meon & Madge, Simon & Quinn, Anthony & Anderson, David & Habib, Nabil. (2010). Vortex keratopathy and fabry disease: a case series highlighting the role of the optometrist. 11.
- Mehta A, Hughes DA. Fabry Disease. 2002 Aug 5 [Updated 2017 Jan 5]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1292/
- Linhart, A., Germain, D.P., Olivotto, I., Akhtar, M.M., Anastasakis, A., Hughes, D., Namdar, M., Pieroni, M., Hagège, A., Cecchi, F., Gimeno, J.R., Limongelli, G. and Elliott, P. (2020), An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur J Heart Fail, 22: 1076-1096. https://doi.org/10.1002/ejhf.1960
- Linhart A. The heart in Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Chapter 20. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11576/
Authors: Michal Pazderník, Vojtěch Berka
.jpg)
You Might Also Like

Eosinophilic Myocarditis Due To Eosinophilic Granulomatosis With Polyangiitis (Churg-Strauss sydrome)
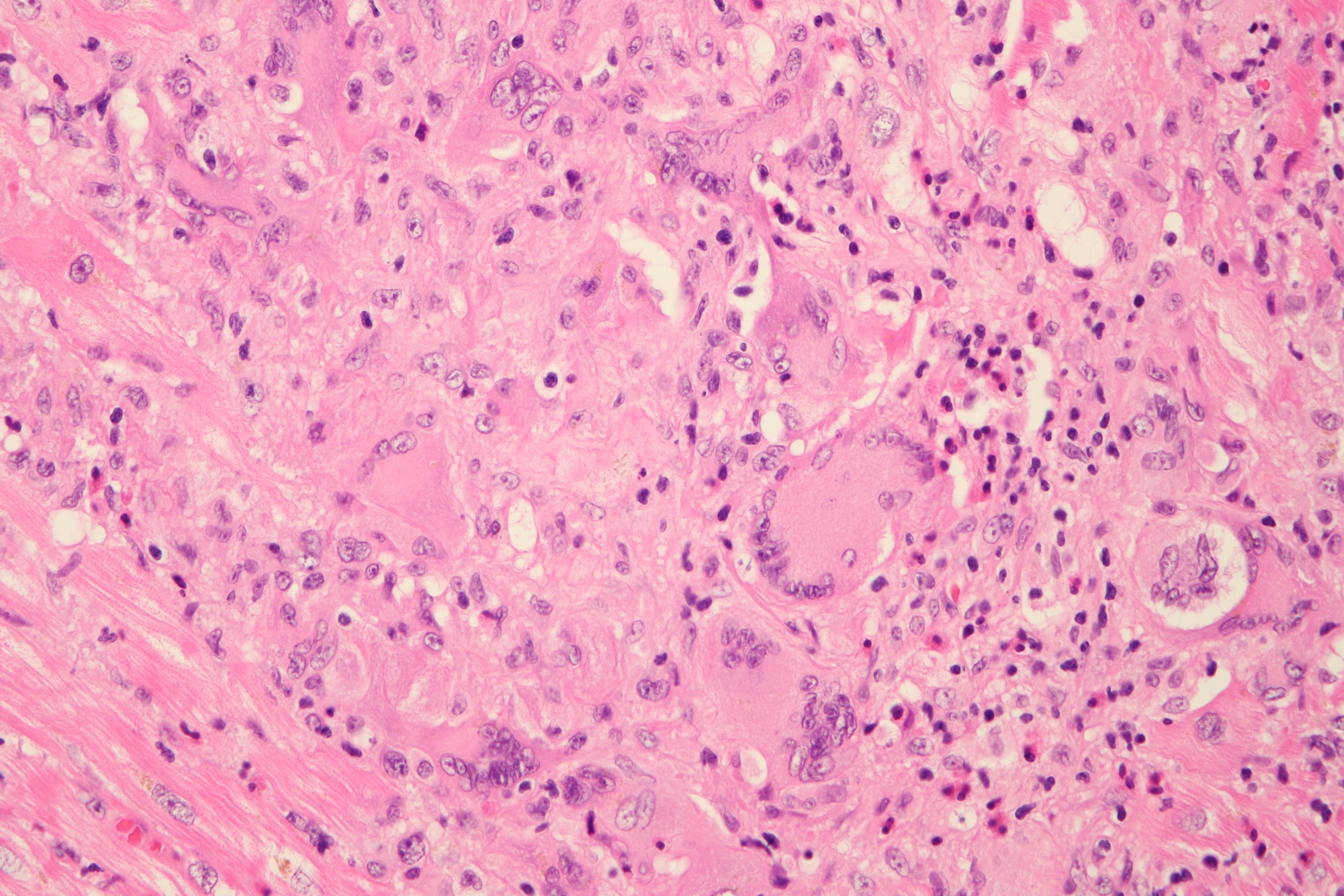
Cardiogenic shock due to Giant Cell Myocarditis